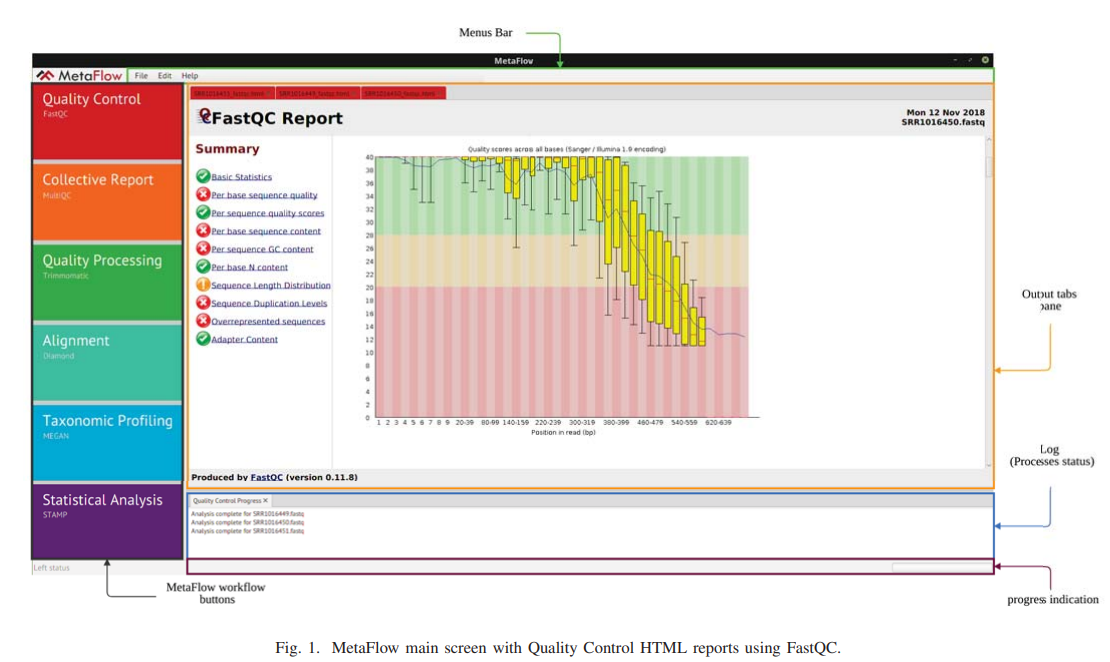
MetaFlow: An interactive user-friendly workflow for automated analysis of whole genome shotgun sequencing metagenomic data
Metagenomics is a rapidly emerging field that is concerned with the study of microbial communities 'microbiomes' on both levels of taxonomic classification and functional annotation. Targeted amplicon (16S rRNA) and whole genome shotgun (WGS) sequencing are the two main sequencing strategies in metagenomics. As amplicon sequencing provides a cheap way to classify the composition of a microbial community, it lacks the ability to identify microbial genes and annotate its corresponding functions. On the other hand, WGS sequencing allows further investigation of the complete genomes with all associated genetic information, and hence, gives another dimension for functional analysis, however, these mentioned advantages are not in favor of sequencing cost, duration of the analysis, and required computational resources. Currently, most tools for WGS microbiome data analysis are commandline based, which limits its use to only experienced bioinformaticians, due to its complexity during installation, usage and fine tuning numerous parameters. Moreover, current tools required for different steps of microbiome data analysis are platform specific and not user-friendly for biologists without previous deep knowledge in command line and installation/configuration troubleshooting abilities. In this paper, we present MetaFlow; a comprehensive automatic platform-independent workflow with a friendly graphical user interface (GUI), for interactive analysis of shotgun metagenomic data. MetaFlow will facilitate the analysis of WGS metagenomic data on a personal laptop, more smoothly, rapidly in an automatic, comprehensive and interactive way for biologists and microbiologists. © 2018 IEEE.